
Carnivorous crocodile-like monsters used to terrorize the Caribbean
How did reptilian things that looked something like crocodiles get to the Caribbean islands from South America millions of years ago? They probably walked.
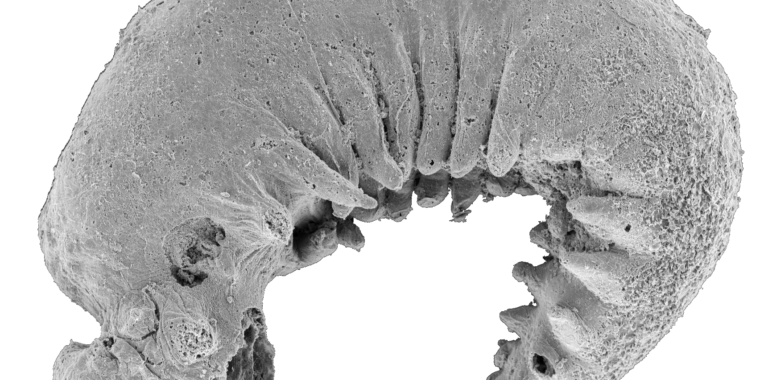
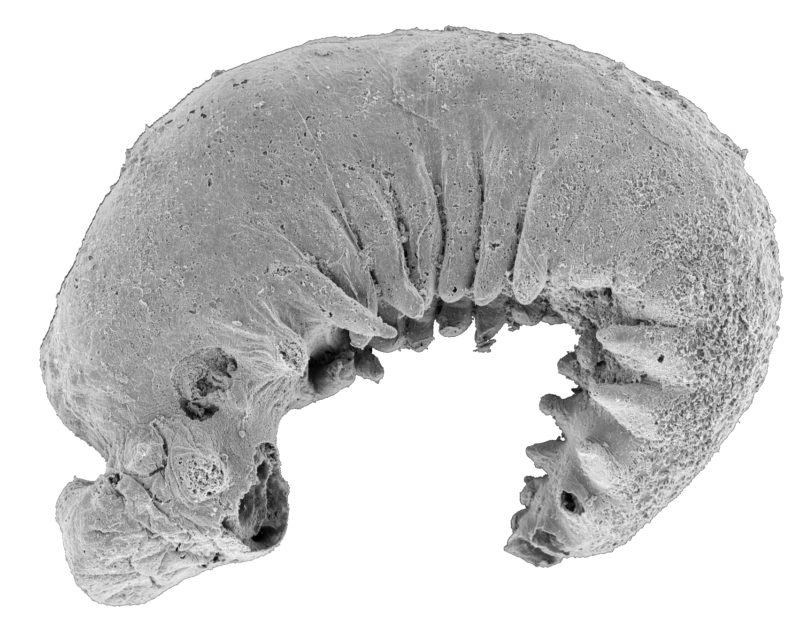
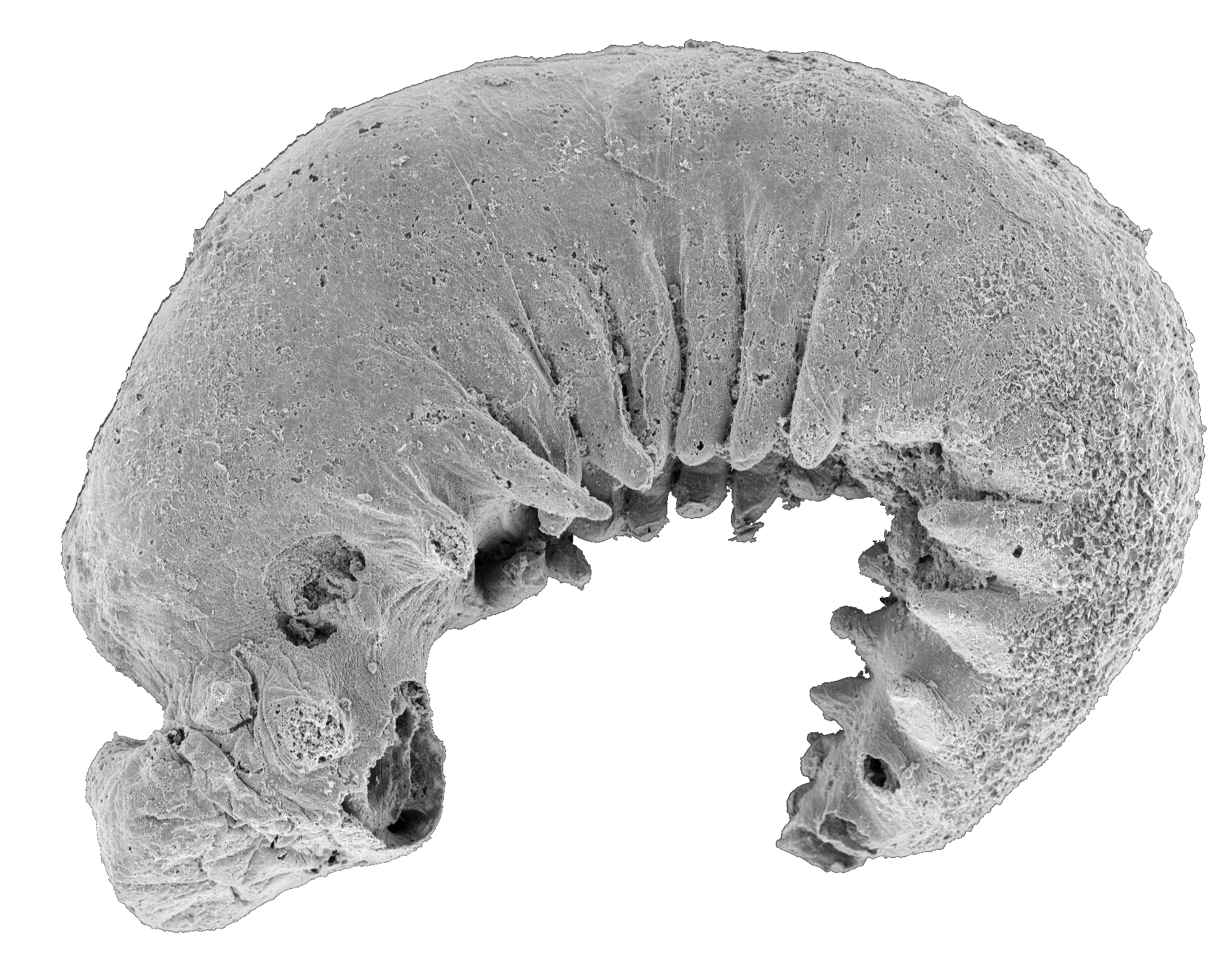
The existence of any prehistoric apex predators in the islands of the Caribbean used to be doubted. While their absence would have probably made it even more of a paradise for prey animals, fossils unearthed in Cuba, Puerto Rico, and the Dominican Republic have revealed that these islands were crawling with monster crocodyliform species called sebecids, ancient relatives of crocodiles.
While sebecids first emerged during the Cretaceous, this is the first evidence of them lurking outside South America during the Cenozoic epoch, which began 66 million years ago. An international team of researchers has found that these creatures would stalk and hunt in the Caribbean islands millions of years after similar predators went extinct on the South American mainland. Lower sea levels back then could have exposed enough land to walk across.
“Adaptations to a terrestrial lifestyle documented for sebecids and the chronology of West Indian fossils strongly suggest that they reached the islands in the Eocene-Oligocene through transient land connections with South America or island hopping,” researchers said in a study recently published in Proceedings of the Royal Society B.
Origin story
During the late Eocene to early Oligocene periods of the mid-Cenozoic, about 34 million years ago, many terrestrial carnivores already roamed South America. Along with crocodyliform sebecids, these included enormous snakes, terror birds, and metatherians, which were monster marsupials. At this time, the sea levels were low, and the islands of the Eastern Caribbean are thought to have been connected to South America via a land bridge called GAARlandia (Greater Antilles and Aves Ridge). This is not the first land bridge to potentially provide a migration opportunity.
Fragments of a single tooth unearthed in Seven Rivers, Jamaica, in 1999 are the oldest fossil evidence of a ziphodont crocodyliform (a group that includes sebecids) in the Caribbean. It was dated to about 47 million years ago, when Jamaica was connected to an extension of the North American continent known as the Nicaragua Rise. While the tooth from Seven Rivers is thought to have belonged to a ziphodont other than a sebacid, that and other vertebrate fossils found in Jamaica suggest parallels with ecosystems excavated from sites in the American South.
The fossils found in areas like the US South that the ocean would otherwise separate suggest more than just related life forms. It’s possible that the Nicaragua Rise provided a pathway for migration similar to the one sebecids probably used when they arrived in the Caribbean islands.
Carnivorous crocodile-like monsters used to terrorize the Caribbean Read More »














{kind=link}
{kind=link}
{kind=link}